Lupus eritematoso sistémico: Aspectos prácticos para el médico de familia
RESUMEN
El lupus eritematoso sistémico (LES) es una enfermedad inflamatoria crónica, multisistémica, de etiología autoinmune, que en el 90% de las ocasiones afecta a mujeres en edad fértil, aunque también se puede presentar en la infancia, en décadas mas tardías y en hombres. Además de los síntomas constitucionales, los síntomas articulares y dermatológicos son los más frecuentes, aunque manifestaciones como serositis, nefritis, citopenia, fenómeno de Raynaud, manifestaciones neurológicas, etc., también son comunes. Cursa con diversas alteraciones de laboratorio, entre las que es característica la presencia de anticuerpos anti-nucleares. La mayoría de los pacientes siguen una evolución crónica y presentan brotes o exacerbaciones de la enfermedad, intercalados con períodos de inactividad. En este post se revisan los criterios diagnósticos, siendo importante su conocimiento por el médico de familia, ya que un diagnóstico precoz se traduce en un mejor pronóstico de una enfermedad potencialmente mortal.
EPIDEMIOLOGÍA
La incidencia y prevalencia del LES varían en función del área geográfica y de la etnia analizada, más elevadas en Europa (en el Reino Unido, la prevalencia estimada es de 28 por 100.000) que en Estados Unidos (5-15 por 100.000 habitantes), siendo mas frecuente y grave en ciertas etnias, como los nativos indígenas americanos, orientales y afroamericanos. Se presenta con mayor con frecuencia entre las edades de 15 y 45 años, es 12 veces más frecuente en mujeres que en hombres.
En España se estima una prevalencia de 34-91 por 100.000 personas, y una incidencia de 2/100.000/habitantes-año.
DIAGNÓSTICO
Los criterios diagnósticos del LES fueron elaborados por el American College of Rheumatology en 1982 y revisados en 1997. Hay que saber que estos criterios fueron desarrollados inicialmente para identificar a los pacientes para estudios clínicos y se basan en una población blanca. Para poder clasificar de LES a un paciente es necesaria la presencia, de forma simultánea o en algún momento durante el curso evolutivo de la enfermedad, de cuatro o más de estos criterios:
- Eritema facial: eritema fijo, plano o elevado sobre eminencias malares.
- Lupus discoide: lesiones cutáneas eritematosas, con cambios en la pigmentación y cicatrices residuales.
- Fotosensibilidad: exantemas causados por exposición a luz UV.
- Úlceras orales: en cavidad oral o nasofaríngea, por lo general indoloras observadas por un médico.
- Artritis: no erosiva, que afecte a dos o más articulaciones periféricas con dolor, inflamación o derrame articular.
- Serositis: pleuritis o pericarditis (ECG), o roce o evidencia de derrame pericárdico.
- Alteración renal: proteinuria > 0,5 g/dL o > 3+ o cilindros celulares o hemáticos.
- Alteración del SNC: convulsiones o psicosis, sin otra causa neurológica
- Alteración hematológica: anemia hemolítica; leucopenia (< 4.000/mm3) o linfopenia (< 1.500/mm3) en 2 o más ocasiones o trombocitopenia (< 100.000/mm3), en ausencia de fármacos que las produzcan.
- Alteración inmunológica: anticuerpos anti-DNAn, anti-Sm y/o anticuerpos antifosfolipídicos.
- Anticuerpos antinucleares: título elevado de anticuerpos ANAs por IFI o ensayo equivalente en algún momento de la evolución, y en ausencia de fármacos que se saben estan asociados a síndrome de lupus inducido por fármacos.
Manifestaciones generales
Astenia, fiebre y pérdida de peso son síntomas frecuentes que se producen en algún momento durante el curso de la enfermedad. La astenia es el más común de éstos, ocurre en el 80% a 100% de los pacientes, pero no se correlaciona con la actividad de la enfermedad. La astenia por otras causas, como la anemia, el hipotiroidismo, medicamentos (por ejemplo, beta-bloqueantes), depresión, fibromialgia y estrés debe ser considerada en el diagnostico diferencial.
La fiebre se presenta en mas del 50% de los pacientes con LES y se asocia a enfermedad activa. Sin embargo, debe excluirse la etiología infecciosa como las infecciones oportunistas relacionadas con el tratamiento inmunosupresor, reacciones a medicamentos, etc.
La pérdida de peso en el LES puede estar relacionada con la actividad de la enfermedad o su tratamiento.
Adenopatias periféricas, más o mensos generalizadas. Las adenopatías son por lo general no dolorosas, varían en tamaño de unos pocos milímetros a 3 a 4 cm y son mas frecuentes en las regiones cervical y axilar. Adenopatías hiliares son poco frecuentes. Los pacientes con linfadenopatía son más propensos a tener manifestaciones constitucionales. El linfoma y la mononucleosis infecciosa deben ser descartados. La histología de las biopsias de los ganglios linfáticos en el LES con frecuencia muestra hiperplasia reactiva.
Manifestaciones mucocutáneas
Las manifestaciones mucutáneas son frecuentes en el LES. El eritema malar “en alas de mariposa”, respetando los surcos nasogenianos, que es fotosensible, se presenta en el 60% de los casos. Puede ser plano o elevado, doloroso y pruriginoso, por lo general dura unos pocos días, se cura sin dejar cicatriz, pero a menudo se repite después de la exposición al sol.
 |
| Añadir leyenda |
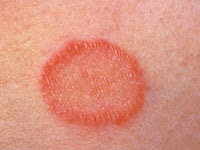
Otras lesiones características son el lupus cutáneo crónico discoide, aparece en el 20% de los casos, se presenta como lesiones con atrofia central, típicamente circulares, con un anillo eritematoso elevado y se sitúan, generalmente sobre cuero cabelludo, pabellones auriculares, cara, zonas de los brazos expuestas al sol y tronco. Estas lesiones terminan dejando cicatriz.

Lupus cutáneo subagudo, caracterizado por lesiones cutáneas eritematosas, fotosensibles, de forma anular o papuloescamoisa. Se suelen localizar en hombros, brazos, cuello y tronco, y curan sin dejar cicatriz. Aproximadamente un 50% de los pacientes con lupus cutáneo subagudo desarrollan LES y un 10% de los LES tienen LECS. Se relaciona con la presencia de anticuerpos anti-Ro/SS-A.+

Úlceras de la boca, y menos frecuentemente úlceras en la nariz, son otra característica de LES, que se presentan en un 12% a 45% de los pacientes. Por lo general son grandes y sin dolor, en contraste con las lesiones herpéticas. Estas úlceras suelen mejorar con simples medidas locales y su curso es paralelo al curso de la enfermedad.
 |
| Añadir leyenda |
Otras lesiones cutáneas y mucosas incluyen: exantemas causados por exposición a luz UV, alopecia, alopecia areata, urticaria, vasculitis eritema multiforme, liquen plano, púrpura, etc.

Manifestaciones musculoesqueléticas
La artritis, acompañada de rigidez matutina, afecta principalmente las articulaciones interfalángicas proximales, metacarpofalángicas, carpo, codos, y rodillas. Tiende a ser simétrica y es típicamente no erosiva. Aunque infrecuentemente, deformidad de las articulaciones puede ocurrir, conociéndose el patrón de deformidad de la articulación en ausencia de enfermedad erosiva como artritis de Jaccoud (desviación cubital y subluxaciones articulares en las manos en la ausencia de daño radiológico). Los pacientes con LES también se puede desarrollar miositis que conduce a la debilidad muscular preferentemente proximal y dolor difuso, acompañado de elevación de la CPK.
Fenómeno de Raynaud
El inicio reciente de cambio de color trifásico (del blanco al azul y después al rojo) en la manos y los pies en una mujer joven debido a la exposición al frío o por emociones, debe motivar la búsqueda de otras características de LES.
Manifestaciones renales
La afectación renal está presente en aproximadamente el 25-50% de los pacientes. Las principales manifestaciones son proteinuria (> 500 mg/24 horas), hematuria y cilindruria, síndrome nefrótico o nefrítico, con o sin insuficiencia renal, HTA. La nefritis lúpica es más frecuente en los pacientes hispanos y negro y en aquellos con enfermedad más grave en otros órganos. Los pacientes con anticuerpos anti-ADNn son más propensos a desarrollar glomerulonefritis.
Manifestaciones neuropsiquiátricas
La frecuencia de manifestaciones del sistema nervioso central en LES oscila entre el 35% y el 75%. Se han identificado una gran variedad de manifestaciones neuropsiquiátricas como cefalea (de características migrañosas o inespecíficas), si bien es controvertida su relación con actividad lúpica, alteraciones del ánimo (ansiedad y depresión), disfunción cognitiva (relacionada con la presencia de lesiones isquémicas cerebrales y anticuerpos antifosfolipídico), síndrome orgánico cerebral agudo lúpico, (cuadros psicóticos, excluidos los asociados a corticoides), enfermedad cerebrovascular y otras manifestaciones menos frecuentes: (convulsiones, parkinsonismos, síndrome desmielinizante, corea, meningitis aséptica, mielopatía, mono o polineuropatía de nervios craneales o periféricos, neuritis óptica, polirradiculoneuritis aguda tipo Guillain-Barré, plexopatía, disautonomía y miastenia gravis).
El diagnóstico de la afectación cerebral en el LES es clínico. Otras causas como sepsis, hipertensión uremia, neoplasias, la epilepsia, miastenia grave, esclerosis múltiple, etc., deben ser excluidas.
Manifestaciones cardiopulmonares
Las manifestaciones cardiovasculares de LES incluyen pericarditis, miocarditis, endocarditis y enfermedad aterosclerótica coronaria. El riesgo cardiovascular global se duplica en los pacientes de cualquier edad con LES y se ve incrementado notablemente en mujeres con edades comprendidas entre 35 y 44 años. La miocarditis se debe sospechar en pacientes con taquicardia, arritmias, defectos de conducción, o cardiomegalia inexplicable.
Las manifestaciones pulmonares del LES incluyen pleuritis, derrame pleural, enfermedad pulmonar intersticial difusa, hipertensión pulmonar y, en raras ocasiones, hemorragia pulmonar. La embolia pulmonar deben ser excluida en los pacientes con LES que presentan dolor torácico pleurítico, disnea, hemoptisis, sobre todo si los anticuerpos antifosfolípido son positivos. Los derrames pleurales en el LES son generalmente unilaterales y en general exudativos.
Manifestaciones hematológicas
Anemia, leucopenia y trombocitopenia son manifestaciones hematológicas frecuentes en el LES. La anemia suele ser secundaria a enfermedades crónicas y mejora con el control de la actividad de la enfermedad. La anemia hemolítica es rara pero puede ser muy grave. La leucopenia es generalmente debido a linfopenia y en menor medida a neutropenia. La trombocitopenia también se observa frecuentemente y otras causas deben ser excluidas. La presencia de anticuerpos antifosfolípidos aumenta el riesgo de trombosis venosa y arterial.
Manifestaciones gastrointestinales
El LES puede afectar a cualquier parte del tracto gastrointestinal. Las úlceras orales son frecuentes. La disfagia es menos frecuente y se debe a alteraciones en la la motilidad esofágica. Dolor abdominal, vómitos y diarrea pueden presentase en el paciente con LES secundarios a peritonitis u oclusión de la arteria mesentérica, pero otras causas de abdomen agudo deben ser excluidas, y aunque es raro, el lupus puede simular una apendicitis. La pancreatitis también puede ser debida a LES, pero es importante excluir como causa subyacente el tratamiento con azatioprina. También la hepatitis crónica activa puede ocurrir en el LES.
Manifestaciones oculares
En el LES se han descrito una gran variedad de manifestaciones oculares: Conjuntivitis, episcleritis, sindrome seco y menos frecuentemente uveítis neuritis óptica vasculitis retiniana, etc.
EXPLORACIONES COMPLEMENTARIAS
Las siguientes pruebas se deben realizar en toda persona con sospecha de LES:
- Hemograma con fórmula y VSG.
- Bioquímica con función renal, perfil hepático, muscular, lipídico, glucemia y PCR. Pruebas de coagulación básicas.
- Orina elemental, con sedimento (hematíes, leucocitos y cilindros) y cuantificación del cociente proteína/creatinina.
- Anticuerpos: FR, ANAs, anti-DNAn, anti-Sm, anti-Ro (SSA), anti-La (SSB), anti-RNP, anticardiolipina IgG-IgM y anticoagulante lúpico.
- Niveles de complemento: C3, C4 y CH50.
- Test de Coombs, si sospecha de hemólisis
- Proteinograma e inmunoglobulinas.
- Mantoux, serología VHB y VHC, previendo vacunaciones y medicación inmunosupresora posterior.
- Rx de tórax y ECG.
Otras exploraciones complementarias
Otras pruebas complementarias, en función de la sintomatología y/o evolución clínica, se deberán solicitar para descartar afectación de distintos órganos y realizar el diagnóstico diferencial con otras entidades:
- Anticuerpos antipéptido cíclico citrulinado (CCP) y radiografía simple de manos: en pacientes que presenten artritis simétrica de pequeñas articulaciones y en los casos de artritis de instauración 1-2 meses antes, o si responde mal al tratamiento, o se sospecha AR.
- Anticuerpos AMA, anti-LKM, anti-músculo liso, cuando exista elevación de enzimas de colestasis y/o citolisis; además de ecografía abdominal, estudio de coagulación. En algunas ocasiones se valorará biopsia hepática.
- Biopsia cutánea en caso de lesión dudosa.
- Biopsia renal si se sospecha glomerulonefritis lúpica.
- Perfil tiroideo: TSH basal y niveles de T4 y T3 libres, anticuerpos antimicrosomales, anti-TPO, ecografía y/o gammagrafía tiroideas.
- Ecocardiograma para valoración de afección valvular, fracción de eyección y cálculo de la presión arterial pulmonar.
- Pruebas de función respiratoria que incluyan difusión del monóxido de carbono si hay sospecha de afección pulmonar; en caso de alteración TAC de tórax de alta resolución.
- RMN cerebral, electroencefalograma, y ocasionalmente punción lumbar y estudio de LCR, en pacientes con manifestaciones del sistema nervioso central.
- Electromiograma cuando se sospeche afectación de sistema nervioso periférico o muscular.
- Determinación de vitamina D y densitometría ósea.
- Prueba de Coombs: deben solicitarse si el recuento sanguíneo inicial muestra anemia hemolítica, como elevación de VCM y del recuento de reticulocitos.
SEGUIMIENTO
Durante su evolución, los pacientes con LES experimentan exacerbaciones, seguidas de periodos de menor actividad o inactividad, y su seguimiento debe individualizarse en cada paciente, dependiendo de la actividad de la enfermedad, de los órganos afectados, del tratamiento utilizado, de la situación personal del paciente, etc., correspondiendo en muchos casos a otras especialidades. Sin embargo, el médico de familia en cada visita debe estar alerta a la aparición de nuevos signos y síntomas clínicos: cutáneos, articulares, serositis, cardiorrespiratorios, neurológicos, etc., así como a síntomas que sugieran efectos adversos derivados del tratamiento. Además el médico de familia debe estar familiarizado con las pruebas más habituales utilizadas en el seguimiento:
- Hemograma.
- Parámetros bioquímicos: glucosa, urea, creatinina, colesterol total, LDL colesterol, HDL colesterol, triglicéridos, GOT/AST, GPT/ALT, GGT, fosfatasa alcalina, CPK, LDH, proteínas totales/albúmina y electrolitos.
- Reactantes de fase aguda: proteína C reactiva (PCR) y velocidad de sedimentación globular (VSG); posible utilidad, sin confirmar, de procalcitonina si se sospecha sobreinfección.
- Sedimento urinario y cociente proteínas/Cr en orina
- Niveles de C3, C4 y anticuerpos anti-DNAn. (No se deben pedir los ANA en las revisiones).
BIBLIOGRAFÍA
- Bernatsky S, Joseph L, Boivin JF, et al. The relationship between cancer and medication exposures in systemic lupus erythematosus: a case-cohort study. Ann Rheum Dis. 2008;67:74-79.
- Bernatsky S. Boivin JF. Joseph L, et al. Race/ethnicity and cancer occurrence in systemic lupus erythematosus. Arthritis Rheum. 2005;53:781-784.
- Bertsias G, Ioannidis JP, Boletis J, et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis. 2008;67:195-205.
- Beyan E, Beyan C, Turan M. Hematological presentation in systemic lupus erythematosus and its relationship with disease activity. Hematology. 2007;12:257-261.
- Breuer GS, Baer A, Dahan D, Nesher G. Lupus-associated pancreatitis. Autoimmun Rev. 2006;5:314-318.
- Cervera R, Abarca-Costalago M, Abramovicz D, et al. Systemic lupus erythematosus in Europe at the change of the millennium: lessons from the "Euro-Lupus Project". Autoimmun Rev. 2006;5:180-186.
- Cervera R, Piette JC, Font J, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002;46:1019-1027.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- D'Cruz DP, Khamashta MA, Hughes GRV. Systemic lupus erythematosus. Lancet. 2007;369:587-596.
- Dubois EL, Tuffanelli DL. Clinical manifestations of SLE. Computer analysis of 520 cases. JAMA. 1964;190:104-111.
- Eaton RB, Schnneider G, Schur PH. Enzyme immunoassay for antibodies to native DNA. Specificity and quality of antibodies. Arthritis Rheum. 1983;26:52-62.
- Estes D, Christian CL. The natural history of systemic lupus erythematosus by prospective analysis. Medicine (Baltimore). 1971;50:85-95.
- Feinglass EJ, Arnett FC, Dorsch CA, et al. Neuropsychiatric manifestations of systemic lupus erythematosus: diagnosis, clinical spectrum, and relationship to other features of the disease. Medicine (Baltimore). 1976;55:323-339.
- Font J, Pallares L, Cervera R, et al. Systemic lupus erythematosus in the elderly: clinical and immunological characteristics. Ann Rheum Dis. 1991;50:702-705.
- Giannakopoulos B, Passam F, Rahgozar S, Krilis SA. Current concepts on the pathogenesis of the anti-phospholipid syndrome. Blood. 2007;109:422-430.
- Ginzler EM, Diamond HS, Weiner M, et al. A multicenter study of outcome in systemic lupus erythematosus. I. Entry variables as predictors of prognosis. Arthritis Rheum. 1982;25:601-611.
- Hallegua DS, Wallace DJ. Gastrointestinal manifestations of systemic lupus erythematosus. Curr Opin Rheumatol. 2000;12:379-385.
- Hochberg MC, Boyd RE, Ahearn JM, et al. Systemic lupus erythematosus: a review of clinico-laboratory features and immunogenetic markers in 150 patients with emphasis on demographic subsets. Medicine (Baltimore). 1985;64:285-295.
- Kaslow RA, Masi AT. Age, sex, and race effects on mortality from systemic lupus erythematosus in the United States. Arthritis Rheum. 1978;21:473-479.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with SLE: comparison with the Framingham study. Am J Epidemiol. 1997;145:408-415.
- Memet B, Ginzler EM. Pulmonary manifestations of systemic lupus erythematosus. Semin Respir Crit Care Med. 2007;28:441-450.
- Nesher G, Breuer GS, Temprano K, et al. Lupus-associated pancreatitis. Sem Arthritis Rheum. 2006;35:260-267.
- Schneebaum AN, Singleton JD, West SG, et al. Association of psychiatric manifestations with antibodies to ribosomal P proteins in systemic lupus erythematosus. Am J Med. 1991;90:54-62.
- Smeenk R, Brinkman K, van den Brink H, et al. Antibodies to DNA in patients with systemic lupus erythematosus: their role in the diagnosis, the follow-up and the pathogenesis of the disease. Clin Rheumatol. 1990;9(suppl 1):S100-S110.
- Sociedad española de medicina interna. Guías clínicas de enfermedades autoinmunes sistémicas: Lupus eritematoso sistémico 2011Disponible en: http://www.chospab.es/biblioteca/libros/GUIA_LUPUS_ERITOMASO.pdf
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271-1277.
- Tan EM, Feltkamp TE, Smolen JS, et al. Range of antinuclear antibodies in "healthy" individuals. Arthritis Rheum. 1997;40:1601-1611.
- Vitali C, Doria A, Tincani A. International survey on the management of patients with SLE. I. General data on the participating centers and the results of a questionnaire regarding mucocutaneous involvement. Clin Exp Rheumatol. 1996;14 Suppl 16:S17-S22.
- von Muhlen CA, Tan EM. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin Arthritis Rheum. 1995;24:323-358.
- Ward MM. Premature morbidity from cardiovascular and cerebrovascular diseases in women with systemic lupus erythematosus. Arthritis Rheum. 1999;42:338-346.
- Weening JJ, D'Agati V, Schwartz MM, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol. 2004;15:241-250.
- Yung RL, Richardson BC. Drug-induced lupus. Rheum Dis Clin North Am. 1994;20:61-86.
- Zandman-Goddard G, Schoenfeld Y. SLE and infections. Clin Rev Allergy Immunol. 2003;25:29-40.
- Zonana-Nacach A, Roseman JM, McGwin G Jr, et al. Systemic lupus erythematosus in three ethnic groups, VI: factors associated with fatigue within five year of criteria diagnosis. Lupus. 2000;9:101-109.


Comentarios
Publicar un comentario
Este es un blog dirigido a profesionales sanitarios. Los comentarios están sujetos a moderación por el autor antes de su publicación, no admitiéndose publicidad, comentarios no profesionales, no fundamentados científicamente, ni aquellos que resulte inapropiados u ofensivos, etc. Tampoco, en ningún caso a través del blog o correo electrónico, se atenderán casos clínicos particulares ni se dará información personalizada. Si algún paciente desea ser atendido en consulta puede solicitar cita en el teléfono indicado para tal fin.