Anemias normocíticas
RESUMEN
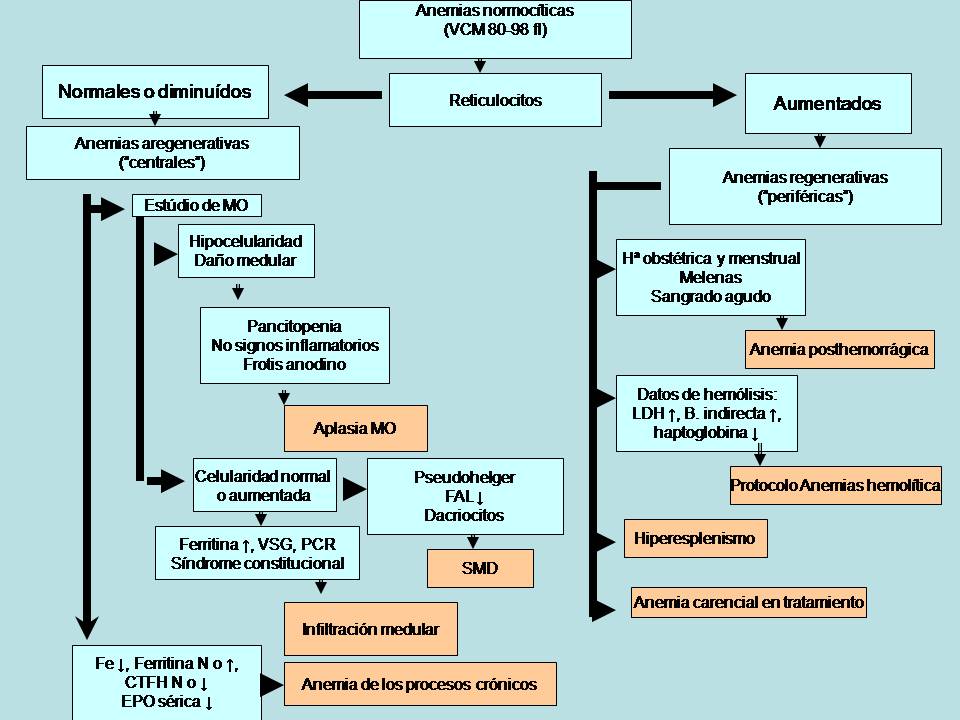
Las anemias normocíticas son un grupo de anemias caracterizadas por presentar un VCM normal (82-98 fl). Frecuentemente suelen tener también la hemoglobina corpuscular media (HCM) normal, y por ello se denominan normocíticas y normocrómicas. La figura 1 muestra un algoritmo diagnóstico de las anemias normocíticas en Atención Primaria.

Figura 1.- Algoritmo diagnóstico de las anemias normocíticas.
B: bilirrubina; CTFH: capacidad total fijadora de hierro; EPO: eritropoyetina sérica; FAL: fosfatasa alcalina leucocitaria; Fe: hierro; LDH: lactatodeshidrogenasa; MO: médula ósea; N: normal; PCR: proteína C reactiva; SMD: síndrome mielodisplásico; VCM: volumen corpuscular medio; VSG: velocidad de sedimentación globular.
La mayor parte de las anemias normocíticas con reticulocitopenia están asociadas con procesos crónicos (insuficiencia renal crónica, artritis reumatoide, neoplasias, conectivopatías, linfomas, etc.), en donde el diagnóstico del proceso subyacente no suele ser generalmente difícil. Sin embargo, se deberán realizar todos los esfuerzos posibles para despistar cualquier otro tipo de anemia asociada (ferropénica, megaloblástica, etc.) que sea susceptible de tratamiento.
La presencia de otras anomalías, como bi o pancitopenia, reacción leucoeritroblástica, etc., son indicación de biopsia de médula ósea para el despistaje de anemia aplasica o anemias por procesos infiltrativos de medula ósea. Los síndromes mielodisplásicos y las alteraciones tiroideas tambien pueden presentarse como anemia normocítica, aunque normalmente se presentan con cierto grado de macrocitosis
La presencia de otras anomalías, como bi o pancitopenia, reacción leucoeritroblástica, etc., son indicación de biopsia de médula ósea para el despistaje de anemia aplasica o anemias por procesos infiltrativos de medula ósea. Los síndromes mielodisplásicos y las alteraciones tiroideas tambien pueden presentarse como anemia normocítica, aunque normalmente se presentan con cierto grado de macrocitosis
Las causas más comunes de anemia normocítica con reticulocitosis son las pérdidas agudas de sangre y la hemólisis. Descartado el sangrado, la historia y la exploración pueden indicarnos la existencia de hemólisis y su causa.
Finalmente, en ocasiones la evaluación diagnóstica de las anemias normociticas puede ser complicada, ya que anemias clásicamente asociadas a microcitosis o macrocitosis pueden presentarse con VCM normal, como la anemia ferropénica o el síndrome mielodisplásico (SMD), respectivamente.
ANEMIAS DE LOS PROCESOS CRONICOS
La anemia asociada a los procesos crónicos constituye la segunda causa de anemia en Atención Primaria, después de la ferropénica y es la anemia más frecuente en el anciano y en pacientes hospitalizados. Se trata de una anemia hiporregenerativa o central. Suele presentarse como una anemia normocítica normocrómica, aunque en el 30% de los casos puede ser macrocítica e hipocrómica.
Su fisiopatología es multifactorial, postulándose los siguientes mecanismos de producción: a) Disminución de la producción de eritropoyetina, así como de la sensibilidad a ella de las células madres eritroides. b) Dificultad para la movilización y el uso efectivo del hierro, tanto de los macrófagos como de los depósitos; esto provoca que, en algunos casos, exista cierto componente de microcitosis. c) Disminución per se de la producción medular de eritroblastos con cifras bajas de hematíes. d) Discreta disminución de la vida media de los hematíes (mediada por hemolisis extracorpuscular)
Etiología
La anemia de los procesos crónicos se asocia a:
- Enfermedades inflamatorias: artritis reumatoide, L.E.S. sarcoidosis, enfermedad inflamatoria intestinal, etc.·
- Insuficiencia renal
- Enfermedades infecciosas: VIH, TBC, sífilis, endocarditis, osteomielitis, pielonefritis, abscesos pulmonares, neumonías, etc
- Neoplasias: linfomas, carcinomas, etc.
- Lesiones tisulares extensas: grandes quemados, úlceras cutáneas, etc.
- Miscelánea: EPOC, hepatopatía alcohólica, neuropatía, endocrinopatías.
Diagnóstico
En el estudio inicial de una anemia normocítica, es fundamental realizar una anamnesis y una exploración física completa como primer paso para la aproximación diagnóstica; las anemias normocíticas son las que más se relacionan con otros trastornos sistémicos (insuficiencia renal, hepatopatía, endocrinopatía, tumores, conectivopatias, etc.), sangrado y hemólisis. Debe interrogarse sobre la presencia de síntomas propios del síndrome anémico e indagar sobre la utilización de fármacos, consumo de drogas de abuso, enfermedades de base o intervenciones quirúrgicas recientes. En la exploración física se deben buscar los signos propios de anemia. La realización de una exploración sistemática debe incluir un tacto rectal para descartar sangrado digestivo, así como identificar signos de posibles patologías asociadas.
La anemias de las enfermedades crónicas cursan con ferropenia, sin embargo en el estudio del metabolismos del hiero, nos encontramos ferritina normal o aumentada, capacidad total de fijación del hierro (CTFH) normal o disminuida. vitamina Otras exploraciones a realizar serán determinación de vitamina B12 y ácido fólico, creatinina sérica, pruebas de función hepática, tiroidea y niveles de cortisol, así como niveles de eritropoyetina (EPO) sérica para descartar déficit de vitamina V12 o acido fólico, hepatopatía, endocrinopatía o insuficiencia renal crónica asociadas. En función de los síntomas predominantesn habrá que realizar las exploraciones oportunas para descartar un tumor, conectivopatia, infección, etc., con una intensidad en el estudio que nos vendrá marcada por la situación funcional del paciente, y en algunas ocasiones será necesario realizar aspirado y biopsia de médula ósea para completar el estudio.
En el caso de la anemia de la insuficiencia renal crónica se pueden producir alteraciones significativas en las cifras de hemoglobina cuando el aclaramiento de creatinina se deteriora por debajo de 40 ml/min. La intensidad de la anemia se relaciona con el grado de insuficiencia renal y puede llegar, con mucha más frecuencia que en otras anemias normocíticas, a provocar síntomas severos.
Tratamiento
El tratamiento de la anemia de los procesos cronicos debe dirigirse hacia las posibles causas tratables de este cuadro.
En general, la intensidad de las anemias normocíticas crónicas, a excepción de la que ocurre en insuficiencia renal terminal y en los síndromes mielodisplásicos, suele ser moderada y no requiere tratamiento específico. Su curso, lentamente progresivo, hace que no se produzca sintomatología, aun con niveles de hemoglobina muy disminuidos.
En aquellos casos en los que la anemia produzca astenia para el nivel de actividad habitual para el paciente o repercusión en otros aparatos y deteriora la calida de vida, debe valorarse la posibilidad de transfusiones repetidas de carácter paliativo o el uso de eritropoyetina. La administración de esta última debe emplearse en casos seleccionados, monitorizando la respuesta y asegurando unas reservas de hierro adecuadas. De forma general, para pacientes con anemia de los procesos crónicos, el aporte extra de hierro oral, en ausencia de déficit asociado del mismo, es una medida perjudicial, tanto por los efectos adversos digestivos que provoca como por el aumento del mismo en un sistema retículoendotelial ya saturado, y sólo es conveniente si coexiste con un déficit real de hierro.
En el caso de la anemia de los procesos crónico asociada a la insuficiencia real, su principal causa es la disminución en la producción de eritropoyetina, aunque también se produce un acortamiento de la vida media del hematíe de origen no aclarado y también se relaciona con discreto sangrado digestivo. Su tratamiento, en el caso de producirse sintomatología, se basa en el aporte exógeno de análogos de eritropoyetina (epoetina alfa o darbepoetina alfa), por vía subcutánea y con controles estrictos. Siempre es necesario descartar, y en su caso corregir, otros factores asociados (déficit de vit B12, hierro, etc.) antes de valorar el tratamiento sustitutivo, y es conveniente garantizar la existencia de depósitos capaces de responder al aumento de necesidades secundario al inicio de la terapia. El tratamiento suele ser eficaz, y permite mantener un hematocrito en torno a 30-35%. El efecto secundario más frecuente suele ser el aumento leve de la tensión arterial.
ANEMIAS HEMOLITICAS
Etiología
Las anemias hemolíticas se producen por cinco mecanismos fundamentales: a) anomalías de la membrana eritrocitaria (ejem. esferocitosis hereditaria); b) déficit enzimáticos eritrocitarios (ejem. déficit de G6PD, PVK) c) hemólisis producida por anticuerpos (ejemplo, anemia hemolítica autoinmune) d) hemoglobinopatias (ejem. talasemias) e) por agentes exógenos: medicamentos (penicilina, metildopa, quinidina); agentes infecciosos (Plasmodium, Mycoplasma); mecánicos (PTT, válvulas cardiacas).
Clínica
La hemólisis intravascular puede presentarse con instauración súbita de anemia, dolor abdominal, hemoglobinuria, con riesgo de la vida del enfermo por CID, shock, insuficiencia renal. La forma crónica sigue un curso silente, produciendo a largo plazo una hemocromatosis por acumulación de Fe en los tejidos.
La hemólisis extravascular suele aparecer de forma crónica, presentando el paciente síntomas moderados de anemia, palidez, ictericia, esplenomegalia y con el paso del tiempo: litiasis biliar, ulceras en las piernas, dolores óseos por eritropoyesis extramedular (cráneo en cepillo). Durante el curso de la enfermedad tambien pueden aparecen: a) crisis hemolíticas agudas por viriasis, embarazo, consumo de habas, o fármacos (déficit de G6PD). b) crisis aplasicas por agotamiento medular
La presencia de ictericia y esplenomegalia en un paciente con anemia deben hacer pensar en anemia hemolítica. El origen étnico ó una historia familiar sugerirá anemia hemolítica congénita. Por contra, un diagnóstico previo de leucemia linfática crónica, linfoma, tumores sólidos, lupus eritematoso sistémico o infecciones por mycoplasma sugerirán anemia hemolítica autoinmune (AHA).
La anemia suele ser normocítica, salvo en los cuadros de hemólisis aguda en los que debido a la marcada reticulocitosis aparece macrocitosis. Aunque el estudio etiológico de una anemia hemolítica generalmente se halla dificultado por el elevado número de causas que pueden producirla, su diagnóstico se realiza con facilidad por la existencia de cinco signos biológicos característicos: a) reticulocitosis; b) hiperregeneración eritroblástica; c) hiperbilirrubinemia no conjugada; d) incremento de la láctico-deshidrogenasa sérica, y e) descenso de la haptoglobina. Los dos primeros pueden observarse también en la hemorragia, pero los tres restantes son indicativos de destrucción eritrocitaria.
Un test de Coombs positivo sugerirá anemia hemolitica autoinmune (AHA) y un test de Ham y de la Sacarosa sugerirán hemoglobinuria paroxística nocturna (HPN). La presencia de esquistocitos sugiere anemia hemolitica microangiopática y la presencia de esferocitos una esferocitosis congénita. En la figura 2 se muestra una aproximación al diagnóstico de las anemias hemoliticas.

ANEMIA APLASICA
La anemia aplasica es un trastorno que se caracteriza por descenso del numero de hematíes, leucocitos y plaquetas en sangre periférica y de sus precursores correspondientes en médula ósea. El inicio de la enfermedad puede ser solapado o brusco, la mayor parte de los pacientes consultan por síndrome anémico y el resto por hemorragia y/o infección. No existen adenopatías ni esplenomegalia, de hecho, la existencia de estas nos hará sospechar que la causa de la pancitopenia es de otro origen.
Las causas más frecuentes son: exposición o contacto con productos tóxicos (benzol, insecticidas, pegamento), radiaciones ionizantes, la administración de fármacos (citostáticos, antiinflamatorios, cloranfenicol, hidantoínas, sales de oro, etc.), algunos procesos infecciosos (hepatitis vírica), el embarazo y muchas de etiología desconocida.
Para establecer el diagnostico es imprescindible la biopsia de medula ósea, donde se encuentra escasa o nula celularidad.
Los tratamientos empleados en la actualidad comprenden: globulina antilinfocítica, trasplante de medula ósea y ciclosporina.
SÍNDROMES MIELODISPLASICOS (SMD).
Los síndromes mielodisplásicos (SMD) constituyen un grupo heterogéneo de desórdenes hematológicos clonales adquiridos, que afectan la célula madre hemopoyética y se caracterizan morfológica y clínicamente por: hematopoyesis ineficaz, progresiva citopenia periférica, displasia en uno o más linajes celulares, médula ósea (MO) hipercelular y displásica con porcentaje variable de blastos, en la mayoría de los casos y tendencia evolutiva a leucemia aguda. En 1982 el grupo Franco-Americano-Británico (FAB) de Hematología clasificó los SMD según criterios morfológicos en sangre periférica y médula ósea. Controversias y desacuerdos obligaron a la OMS en 1999 a hacer una revisión de la misma y propone la nueva clasificación. La OMS introduce la importancia no sólo de las características morfológicas, sino también la relevancia de la clínica, de la citogenética, el inmunofenotipo y la información biológica para poder definir las distintas entidades. La clasificación OMS se correlaciona mejor con el pronóstico, respuesta terapéutica y progresión a Leucemia Aguda que la clasificación FAB.
Anemia refractaria (AR), 10-20% de los SMD
· Frotis SP: anemia, <1% de blastos
· Médula ósea: displasia eritroide (en ≥ 10% de las células), <5% de blastos.
· La transformación a leucemia aguda es poco común y la supervivencia media varía entre 2 y 5 años en la mayoría de series.
Neutropenia refractaria (RN), <1% de todos los SMD
· Frotis sanguineo: neutropenia, <1% de blastos
· Médula ósea: displasia granulocítica unilineage, blastos <5%
Trombocitopenia refractaria (RT); <1% de todos los SMD
· Frotis sanguineo: trombocitopenia, <1% de blastos
· Médula ósea: displasia megacariocítica unilineage, blastos <5%
Anemia refractaria con sideroblastos en anillo (ARSA), 11.3% de todos los SMD
· Frotis sanguineo: anemia sin blastos
· Médula ósea: displasia eritroide, blastos <5%, ≥ 15% de sideroblastos en anillo.
· El pronostico es similar a AR. Aproximadamente del 1% al 2% de los casos de ARSA se convierten en leucemia mieloide aguda.
Citopenia refractaria con displasia en múltiples líneas (RCMD), el 30% de todos los SMD
· Frotis sanguineo: citopenia (s), blastos <1%, no bastones de Auer
· Médula ósea: displasia multilineaje ± sideroblastos en anillo, blastos <5%, no bastones de Auer
· La supervivencia media general es de 33 meses.
Anemia refractaria con exceso de blastos, tipo 1 (AREB-1), el 40% de todos los SMD
· Frotis sanguineo: citopenia (s), blastos <5%, no bastones de Auer
· Médula ósea: displasia mono o multilinaje, blastos 5-9%, sin bastones de Auer
· Aproximadamente el 25% de los casos de AREB-1 evolucionan a LMA. La supervivencia media es de aproximadamente 18 meses.
Anemia refractaria con exceso de blastos, tipo 2 (AREB-2)
· Frotis sanguineo: citopenia (s), blastos 5-19%, ± bastones de Auer
· Médula ósea: displasia mono o multilinaje, blastos de 10-19%, ± bastones de Auer
· Aproximadamente el 33% de los casos de AREB-2 evolucionan a LMA. La supervivencia media para el AREB-2 es de aproximadamente 10 meses.
SMD relacionado con anomalía cromosómica 5q31 (deleción del cromosoma 5q), poco frecuentes
· Frotis sanguineo: anemia, recuento de plaquetas normal o aumentado, blastos <1%
· Médula ósea: supresión del cromosoma 5q31, blastos <5%
SMD de la infancia, incluyendo citopenia refractaria de la infancia (RCC), <1% de todos los SMD
· Frotis sanguineo: pancitopenia
· Médula ósea: blastos <5%, médula hipocelular
SMD sin clasificar (MDS-U)
· Frotis de sangre periférica: citopenias, blastos ≤ 1%, no Bastones de Auer
· Médula ósea: no se ajusta a ninguna de las categorías anteriores, blastos <5%, no bastones de Auer
Quedaron excluidos de esta clasificación la leucemia mielomonocítica crónica (LMMoC) y la anemia refractaria con exceso de blastos en transformación (AREB-t); la LMMoC debido a su heterogeneidad y su estrecha relación con los procesos mieloproliferativos. La OMS incluye a la LMMoC, junto con la leucemia mieloide crónica atípica (LMCa) y las formas juveniles de LMMoC, LMMJ, en un nuevo grupo llamado Síndromes Mielodisplásicos/ Síndromes Mieloproliferativos (SMD/SMP). La AREB-t fue excluida de los SMD e incluida entre las leucemias agudas mieloblásticas (LAM) tipo M2 (con maduración), debido a que esta nueva clasificación establece que la presencia de más de 20% de blastos en MO es una leucemia aguda. Además las características biológicas y el tratamiento de AREB-t son similares a la de la LAM.
Los SMD pueden ser idiopáticos o secundarios a radiación, quimioterapia, exposición a sustancias tóxicas. Las formas de presentación clínica fluctúan desde citopenias leves sin síntomas, hasta pancitopenias graves. Puede producirse progresión hasta insuficiencia medular o leucemia aguda. Las anemias sideroblásticas constituyen un grupo heterogéneo de trastornos hereditario o adquiridos caracterizados por un metabolismo alterado del Fe de los hematíes. Dentro de las causas de las formas adquiridas se incluyen: isoniacida, cloranfenicol, etanol, saturnismo, neoplasias, inflamación en infecciones crónicas. La anemia sideroblástica adquirida idiopática se considera un SMD.
Respecto al tratamiento la esperanza moderna, aparte del transplante de médula ósea, lo constituye la terapia génica.
Si te ha interesado este post también te puede interesar Curso Online de Hematología para Atención Primaria
Bibliografía:
- Arndt PA, Garratty G. The changing spectrum of drug-induced immune hemolytic anemia. Semin Hematol. 2005;42:137-144.
- Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70:177-182.
- Bain BJ. Diagnosis from the blood smear. N Engl J Med. 2005;353:498-507.
- Bladé JS, Desramé J, Corberand D, Lecoules S, Blondon H, Carmoi T, et al. Diagnosis of anemia in alcoholic cirrhosis. Rev Med Interne. 2007;28:756-65.
- Brodsky RA, Jones RJ. Aplastic anaemia. Lancet. 2005;365:1647-1656.
- Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
- Héctor Rodríguez J y Del Luján Acosta I. Actualización en síndromes mielodisplásicos (SMD). Rev. Méd. Rosario 77: 24-41, 2011. Disponible en: http://www.cimero.org.ar/pdf/VOL77/Rodriguez.pdf
- Jelkmann W. Developments in the therapeutic use of erythropoiesis stimulating agents. Br J Haematol. 2008;141:287-297.
- Keohane EM. Acquired aplastic anemia. Clin Lab Sci. 2004;17:165-71.
- Marsh JC, Ball SE, Darbyshire P, et al; British Committee for Standards in Haematology. Guidelines for the diagnosis and management of acquired aplastic anaemia. Br J Haematol. 2009;147:43-70.
- Özatli D, Köksal AS, Hazneda-Roglu IC, Simsek H, Karakus S, Büyükasik Y, et al. Erythrocytes: anemias in chronic liver diseases. Hematology. 2000;5:69-76.
- Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699-3709.
- Ribes E. Anales de cirugía cardiaca y vascular. 2004;10(1):8-76.
- Rizzo JD, Brouwers M, Hurley P, et al. American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update on the use of epoetin and darbepoetin in adult patients with cancer. J Oncol Pract. 2010;6:317-320.
- Roy CN. Anemia of inflammation. Hematology Am Soc Hematol Educ Program. 2010;2010:276-280.
- Scrijvers D, Roila F; ESMO Guidelines Working Group. Erythropoiesis-stimulating agents in cancer patients: ESMO recommendations for use. Ann Oncol. 2009;20(suppl 4):159-161.
- Tefferi A. Anemia in adults: a contemporary approach to diagnosis. Mayo Clin Proc. 2003;78:1274-80.
- Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med 2005; 352: 1011-23.
- Young NS. Acquired aplastic anemia. Ann Intern Med. 2002;136:534-546.


{kind=link}
{kind=link}
Comentarios
Publicar un comentario
Este es un blog dirigido a profesionales sanitarios. Los comentarios están sujetos a moderación por el autor antes de su publicación, no admitiéndose publicidad, comentarios no profesionales, no fundamentados científicamente, ni aquellos que resulte inapropiados u ofensivos, etc. Tampoco, en ningún caso a través del blog o correo electrónico, se atenderán casos clínicos particulares ni se dará información personalizada. Si algún paciente desea ser atendido en consulta puede solicitar cita en el teléfono indicado para tal fin.